3. УСПЕХИ ХИМИИ И ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
Влияние достижений химии на развитие химической технологии
Промышленный переворот второй половины XVIII - начала XIX в., приведший к интенсивному развитию машинной техники и росту крупных предприятий, вызвал в ряде европейских стран большой спрос на химическую продукцию, стимулировал создание химических заводов. Это, в свою очередь, способствовало развитию химической науки. Химия и физика с середины XVIII в. хотя еще и робко, но все же начинают влиять на практику в отличие от предыдущего периода, когда наука только собирала и обобщала данные практики.
Важнейшей проблемой в химии являлось развитие атомно-молекуляр-ного учения, неразрывно связанное с утверждением основных научных понятий химии: атома, молекулы, валентности - и открытием важнейших законов.
В начале XIX в. выдающиеся ученые Д. Дальтон, А. Авогадро, И. Я. Берцелиус, М. Фарадей, Г. II. Гесс и другие внесли в химию и физику новую живительную струю [1, 2].
Английскому ученому Д. Дальтону принадлежат основополагающие работы по химической атомистике. Руководствуясь атомной гипотезой, Дальтон вывел (1803 г.) один из основных законов, доказав его правомерность в опытном порядке на примере углеводородов. На основе этого закона он составил таблицу «атомных весов» атомов элементов и «сложных атомов» соединений.
Ф. Энгельс в «Диалектике природы», высоко оценив работы Д. Дальтона, назвал его «отцом современной химии», отметив, что «новая эпоха начинается в химии с атомистики...» (Маркс К., Энгельс Ф. Соч., т. 20, с. 608). Вместе с тем в учении Д. Дальтона еще не было правильного представления о различиях между атомом и молекулой, которую он называл сложным атомом. Он считал, что простые газы состоят не из молекул, а из атомов. В своих теоретических исследованиях Д. Дальтон пользовался новыми знаками химических элементов, обозначавшими одновременно и атомные веса.
В 1811 г. итальянский ученый А. Авогадро выдвинул гипотезу, согласно которой молекулы простых газов состоят из одного или нескольких атомов. А. Авогадро представил молекулу как наименьшую частицу простого или сложного вещества, вступающую в химическое взаимодействие. На основе своей гипотезы ученый дал формулировку одного из основных законов идеальных газов. Согласно закону Авогадро в равных объемах различных газов при одинаковых температурах и давлениях содержится одинаковое число молекул [3, с. 181-189]. Закон Авогадро позволил определить молекулярный вес любого вещества. Зная молекулярный вес и химический состав соединений, можно было вычислить атомные веса элементов.
Однако из-за господствовавшего в науке первой половины XIX в. смешения понятий атома, эквивалента и молекулы закон Авогадро не был принят большинством химиков и физиков.
Окончательное разграничение понятия атома и молекулы принадлежит итальянскому химику С. Канниццаро, который на основе закона П. Л. Дюлонга и А. Пти (1819 г.) уточнил значение атомных весов некоторых элементов и показал всеобщую применимость закона Авогадро для определения молекулярных весов простых и сложных веществ в парообразном состоянии. Свои взгляды С. Канниццаро изложил в публикациях (1858 г.) и в докладе на Международном конгрессе химиков, который состоялся в 1860 г. в Карлсруэ (Германия). После этого его работы получили всеобщее признание [3, с. 211-227].
Развитию химии в первой половине XIX в. способствовала деятельность шведского химика и минералога И. Я. Берцелиуса, экспериментально обосновавшего атомистическое учение. Признавая реальность атомов и возможность познать природу химических соединений, ученый своими трудами много способствовал утверждению атомистики и ее внедрению в химии. В 1810-1816 гг. И. Я. Берцелиус дал новые доказательства закона кратных отношений - основного закона химии, открытого Д. Дальтоном еще в 1803 г. В 1814 г. Берцелиус по результатам своих исследований составил таблицу атомных весов 41 элемента, заменив (1811 г.) дальтоновские знаки атомов элементов начальными (одной или двумя) буквами их латинских и греческих названий.
Введенные Берцелиусом символы химических элементов сохранились до наших дней. В 1812-1819 гг. он выдвинул электрохимическую теорию, которая была прогрессивной в истории химии [4, 5].
Расширение знаний об электричестве и применение электрического тока для разложения сложных химических веществ привели в начале XIX в. к новым открытиям в области химии. В 1800 г. английские химики В. Пикольсон и А. Карлейль построили вольтов столб из 17 гальванических элементов и разложили воду на водород и кислород. Затем Г. Дэви (Англия) открыл элементы - калий, натрий, кальций, барий, стронций и магний, разлагая электрическим током их соединения.
В 1834 г. английский физик М. Фарадой открыл закон электролиза, в соответствии с которым количество вещества, выделившегося при электролизе, прямо пропорционально количеству электричества, прошедшего через электролит. Открытие этого закона, а также другие работы М. Фарадея по электролизу заложили основы электрохимии, предложенная им терминология сохранилась до наших дней.
Русский химик, академик Петербургской академии наук Г. И. Гесс в 1840 г. сформулировал основной закон термохимии, согласно которому при химическом процессе всегда выделяется одно и то же количество тепла независимо от того, протекает процесс в одну или в две и более стадий. Правда, в то время основные положения этого закона не нашли поддержки в широких научных кругах, не сумевших оценить значение, которое могло иметь это открытие для развития химии.
В 1852 г. английский ученый Э. Франкленд предложил понятие валентности, внесшее упорядоченность в представление о химических соединениях. Развитие учения о валентности вскоре нашло отражение в трудах химиков-органиков Ф. А. Кекуле (Германия), А. С. Купера (Шотландия) и некоторых других. Однако все они не видели связи между строением молекулы и свойствами вещества.
Эту задачу блестяще осуществил русский химик А. М. Бутлеров, труды которого (1861 г.) легли в основу классической теории химического строения [6]. Бутлеров впервые объяснил явление изомерии, характеризующееся существованием веществ с одинаковым элементарным составом и молекулярным весом, но отличающихся по химическому строению, а также химическим и физическим свойствам.
Теория химического строения Бутлерова, работы немецкого химика Ф. А. Кекуле открыли широкие возможности для органического синтеза. Большой вклад в развитие синтеза органических соединений внесли также французский химик М. Бертло, русский химик В. В. Марковников и ряд других.
Следует отметить, что к проблеме синтеза органических соединений было привлечено внимание ученых еще в первой половине XIX в. Начало было положено работами немецкого химика Ф. Вёлера, впервые синтезировавшего в 1824-1828 гг. из неорганических веществ органическое соединение, напоминающее мочё*вину. В 1842 г. русский химик Н. Н. Зи-нин синтезировал анилин и ряд других веществ. Его работы стали основополагающими в создании промышленности синтетических красителей, фармацевтических препаратов и взрывчатых веществ. Немецкий химик II. П. Грисс в 1858 г. открыл реакцию диазотирования, впервые получив азокрасители [7, 8].
Крупнейшим достижением химии начала второй половины XIX в. явилось открытие в 1869 г. Д. И. Менделеевым периодического закона химических элементов. Этим фундаментальным законом было установлено периодическое изменение свойств химических элементов и образуемых ими «простых и сложных тел», находящихся в периодической зависимости от их атомного веса [9]. Периодический закон позволил обнаружить закономерности свойств химических соединений различных элементов. Периодический закон и созданная на его основе периодическая система элементов Д. И. Менделеева сыграли важнейшую роль в дальнейшем изучении структуры атома, радиоактивности, в открытии изотопии. Появилась возможность рассматривать все элементы в их взаимной связи и прогнозировать свойства неизвестных элементов [10].
Как видим, рассматриваемый период оказался исключительно плодотворным в развитии химии, достижения которой все более и более влияли на технологию химического производства. О химической технологии как о науке впервые стали упоминать в 70-х годах XVIII в. В 1772 г. профессор Геттингенского университета И. Бекман дал определение понятия химической технологии.
Особенно быстро стала развиваться химическая технология после французской буржуазной революции 1789 г. Покончив с феодально-абсолютистским строем во Франции и обеспечив установление в стране буржуазных производственных отношений, французская буржуазная революция способствовала утверждению капитализма в Европе. Буржуазия стремилась разрушить старый социально-общественный строй, устранить преграды, стоящие на пути прогресса производительных сил. В результате сложились благоприятные условия для развития производства, а также технического образования. Сначала во Франции, а затем и в других странах были учреждены первые высшие школы для подготовки инженеров и техников, где усилилось преподавание математики, естественных и технических наук. В Национальном институте Франции (до 1791 г.- Академия наук), объединившем лучшие научные силы страны, были решены некоторые крупные вопросы в области химии и химической технологии; ставилась задача освободить французскую экономику от иностранной зависимости.
Среди учебных заведений, в которых преподавалась химическая технология, необходимо отметить Политехнический институт во Франции, основанный в 1794 г. В 1815 г. был создан Политехнический институт в Вене, в 1821 г.- Ремесленная академия в Берлине [11, с. 16]. В России старейшими высшими школами, которые подготавливали химиков-технологов, были Практический технологический институт, созданный в 1828 г. (ныне Ленинградский технологический институт им. Ленсовета), Московское ремесленное училище «для подготовки искусных мастеров с теоретическими сведениями», основанное в 1830 г. Кафедры химической технологии существовали с начала XIX в. в большинстве университетов. Химическая технология как самостоятельная научная дисциплина была представлена в конце XVIII - начале XIX в. в Петербургской академии наук.
В этот же период создаются первые учебные пособия по химической технологии, а также различные руководства, в которых этой области знаний посвящались самостоятельные разделы. Так, в России профессор Московского университета И. А. Двигубский издал учебник «Начальные основания технологии...» в двух частях [12], вышедший в свет в 1807- 1808 гг. В 1828 г. русский ученый Ф. А. Денисов издал учебник «Пространное руководство к общей технологии или к познанию всех работ, средств, орудий и машин, употребляемых в разных технических искусствах». В этом учебном пособии автор впервые выделяет общую часть, посвященную химическим процессам и аппаратам.
Общий подъем науки в конце XVIII - начале XIX в., учреждение в это время специальных высших учебных заведений и школ, в которых важнейшими научными дисциплинами были химия и химическая технология, определялись запросами промышленности. Промышленный переворот привел в движение практически все отрасли производства, особенно текстильную и металлургическую, сыгравшие огромную роль в развитии химических производств. Увеличился спрос на сырые материалы. Наряду с ростом продукции добывающей промышленности, в общем объеме производимых сырых материалов возросло значение химических продуктов. Их производство обуславливалось стремлением заменить дефицитные виды традиционного сырья более дешевыми и доступными химическими материалами; используются «суррогаты» и отходы производства (См.: Маркс К., Энгельс Ф. Соч., т. 25, ч. I, с. 131-132).
В развитии химических производств отчетливо выявились основные направления, связанные с изысканием новых сырьевых источников для производства соды, серной кислоты, более эффективных белящих, протравных и красящих веществ, необходимых для текстильной, металлообрабатывающей, стекольной, кожевенной, жировой и других отраслей промышленности.
Крупнейшими вехами в развитии мировой химической промышленности в рассматриваемый период были: разработка производства соды по методу Н. Леблана (1791 г.), серной кислоты камерным способом (1746 г.), суперфосфата (1840 г.), соды аммиачным способом Э. Сольве (1863 г.) [13]. Была выдвинута идея получения серной кислоты контактным методом (1831 г.).
Прогресс в области технологии содового производства
Разработка производства соды во Франции по методу Н. Леблана оказала исключительно большое влияние на развитие химической промышленности многих стран. К проблеме получения соды искусственным путем научная мысль обращалась и до Леблана, однако попытки поставить ее производство на промышленную основу были безрезультатными. Потребность различных производств в соде быстро возрастала. Издавна соду добывали, сжигая щелочесодержащие растения. Производство растительной соды особенно сильно развилось на средиземноморском побережье Испании, а также во Франции и в меньших размерах в Шотландии. Испанская сода значительно превосходила по качеству растительную соду Франции и Шотландии.

Производство соды по Леблану (XIX в.)
Особенно ценилась аликантбкая сода, иначе называемая «бариллой». Бариллу получали из специально разводимого на побережье Средиземного моря растения «Solsola soda», зола которого содержит значительные количества соединений натрия, в отличие от других растений, более богатых соединениями калия (поташ). Поле, на котором сеяли это растение, вспахивали три раза в году, а после всходов несколько раз поливали морской водой. Через пить месяцев, когда растения созревали, их извлекали из земли и складывали на сухую землю для просушки, затем загружали в яму в 4 фута шириной и 3 фута глубиной. Здесь проводили сжигание, продолжавшееся около суток. В результате получали сухой, почти стекловидный остаток, который делили на большие куски и транспортировали тюками весом до 400-500 фунтов. Вполне понятно, что при такой примитивной обработке растений состав продукта был непостоянным и колебался в довольно широких пределах. Содержание карбоната натрия (соды) в высшем сорте бариллы составляло 25-30%, а в среднем и низшем сорте 14-20% [14, с. 1-13].
Над проблемой искусственного получения соды Н. Леблан работал с 1787 по 1789 г. В результате ему удалось разработать первый промышленный способ производства соды из поваренной соли. В основу процесса было положено взаимодействие поваренной соли с серной кислотой. Получаемый полупродукт - сернокислый натрий (глауберова соль) - затем обрабатывали в специальных печах с углем и углекислым кальцием. В 1791 г. Н. Леблан получил патент на свой способ, и в том же году во Франции начал работать первый завод по его схеме [ 14, с. 30-31].
Значение содового производства в революционной Франции, скованной блокадой, было особенно велико, так как молодая французская республика была лишена возможности приобретать в других странах селитру, необходимую для производства пороха. Следовало внутри страны высвободить максимальное количество поташа, потребляемого стекольной, мыловаренной и другими отраслями промышленности, где он мог быть заменен содой. Не случайно, что в ответ на изданное в один из наиболее опасных моментов для Франции (1794 г.) постановление Комитета общественного спасения о необходимости разрешить содовую проблему и связанную с ней «фабрикацию селитры» в Комиссию было подано около 30 предложений [14, с. 34-35]. Лучшим способом получения соды искусственным путем был признан способ Н. Леблана.
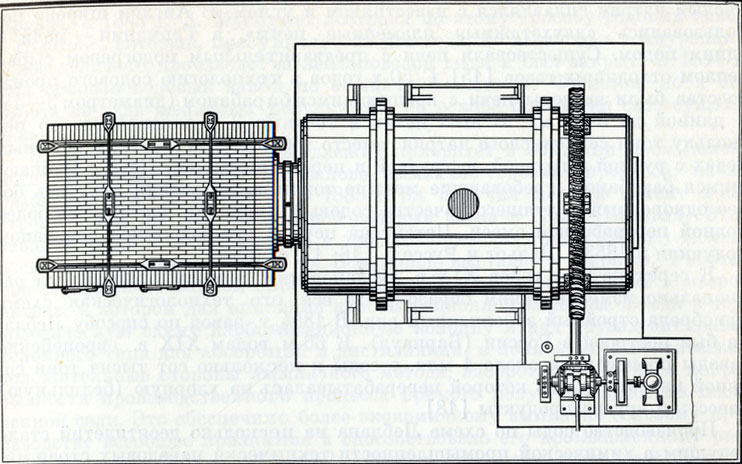
Схема механической содовой печи с вращающимся барабаном
Однако ввиду политических событий во Франции и длительных войн содовая промышленность не получила в первое время достаточного развития. В связи с небольшими масштабами производства цены на искусственную соду оставались весьма высокими, приближающимися к ценам на испанскую соду. До начала второй четверти XIX в. содовое производство не вышло за пределы Франции.
Во Франции к началу 20-х годов XIX в. затраты поваренной соли для производства соды по методу Леблана уже составили 40 тыс. т, что соответствовало 25-30 тыс. т соды стоимостью 2-3 млн. франков. В это время внутреннее производство в ценностном выражении стало равно ввозу. Стоимость искусственной соды уже была ниже испанской и за 20 лет снизилась почти в десять раз: с 800-1000 франков до 100 франков за 1 т. Спустя 10 лет Франция стала ввозить всего лишь 6- 7 тыс. т соды [14, с. 38].
Со второй четверти XIX в. и в последующие десятилетия содовое производство по способу Леблана широко распространяется во всех экономически передовых странах Европы, претерпевая технические усовершенствования и достигая больших успехов в расширении ассортимента выпускаемых продуктов:
К числу крупнейших усовершенствований относится создание закрытых пламенных печей, позволивших утилизировать получающуюся в процессе перевода поваренной соли в сернокислый натрий соляную кислоту. В 1836 г. Госсаж получил привилегию на закрытую пламенную печь, которую в 1839 г. усовершенствовал Гамбль. Продолжительное время на второй стадии производства, связанной с превращением сернокислого натрия в «сырую» соду, применяли пламенные печи, в которых серно-кислый натрий сплавлялся с известняком и углем. В Англии широко использовались «двухэтажные пламенные печи», в Германии - печи с одним подом. Существовали печи с предварительным подогревом сырья теплом отходящих газов [15]. С 50-х годов в технологию содового производства были введены печи с вращающимся барабаном (диаметром 3-4м и длиной до 5-9 м). В этих печах в течение 2 ч сплавлялось по нескольку тонн сернокислого натрия вместо 150 кг, получаемых в обычных печах с ручной загрузкой, выгрузкой и перемешиванием. Печи с вращающимся барабаном, требовавшие меньше топлива, позволяли получать более однородный и лучшего качества содовый плав, что достигалось более полной переработкой смеси. Патент на печи с вращающимся барабаном получили в 1853 г. Элльот и Руссель [16; 17, с. 296].

Общий вид механической содовой печи с вращающимся барабаном
К середине 70-х годов XIX в. леблановский содовый процесс был рационально изменен таким образом, что вся его технологическая схема приобрела стройный законченный вид. В 1864 г. завод по способу Лебла-на был построен в России (Барнаул). К 60-м годам XIX в. европейские заводы производили около 1 млн. т соды и несколько сот тысяч тонн соляной кислоты, часть которой перерабатывалась на хлорную (белильную) известь и другие продукты [18].
Производство соды по схеме Леблана на несколько десятилетий стало ведущим в химической промышлепности технически передовых стран мира. Содовые заводы, работавшие по леблановскому процессу, постепенно разрастались в крупные комбинаты. Однако это производство было сопряжено с рядом трудностей эксплуатационного характера: способ требовал многих видов сырья и давал сравнительно дорогую продукцию. В результате развернулись поиски новых, более экономичных способов получения соды.
Задачу получения соды по новой, аммиачной технологической схеме удалось успешно разрешить в 1863 г. бельгийскому инженеру Э. Сольве. В основу его процесса была положена ставшая известной еще в первых десятилетиях XIX в. схема, исходившая из реакции обменного разложения хлористого натрия и бикарбоната аммония. Получаемый в результате химической реакции бикарбонат натрия последующим прокаливанием переводили в карбонат натрия (соду); другой продукт этой реакции - хлористый аммоний - сначала при помощи извести перерабатывали в аммиак, а затем углекислым газом снова переводили в бикарбонат аммония; в отходах оставался хлористый кальций.
Внимание ученых и инженеров много лет привлекала обменная реакция между хлористым натрием и бикарбонатом аммония, т. е. основная реакция будущего аммиачно-содового процесса. Есть предположение, что эту реакцию многие химики неоднократно открывали независимо друг от друга.
Однако несмотря на кажущуюся простоту обменной реакции между хлористым натрием и бикарбонатом аммония, использование ее в схеме технологического процесса фабричного производства оказалось делом весьма сложным. Об этом свидетельствуют усилия английских специалистов Грей Дьюара п Дж. Хемминга, получивших в 1838 г. первый, а в 1840 г. второй патент на производство соды по аммиачному способу. В их патентах намечены в последовательном порядке все основные операции современного аммиачно-содового процесса: абсорбция, карбонизация, фильтрация, кальцинация и дистилляция. Однако техническое несовершенство аппаратуры помешало реализовать схему в промышленности. Правда, в 1848 г. Д. Муспратт построил по этому способу содовый завод в Ньютоне, который, просуществовав два года, не выдержал конкуренции более дешевого производства леблановской соды и был закрыт. В 1854 г. был основан содовый завод по аммиачной схеме во Франции, но и его работа не дала желаемых результатов: он вскоре прекратил свое существование.
Э. Сольве, которому принадлежит приоритет в создании промышленного пронзводства соды на основе аммиачно-содового процесса, начал разрабатывать его в 1861 г. В 1863 г. он получил первый патент и построил в том же году небольшой содовый завод в Куйе (Бельгия). Этот завод вступил в эксплуатацию в 1865 г. Через два года (1867 г.) Сольве экспонировал образцы соды на Всемирной выставке в Париже. Работая над совершенствованием своего процесса, Э. Сольве взял в 1872 г. второй патент, в котором дал описание карбонизационной колонны. Введя в технологическую схему карбонизационную колонну и ряд других аппаратов колонного типа для абсорбции и дистилляции, в полной мере использовав положительные стороны аммиачного метода, изобретатель добился непрерывности производственного процесса прямого получения соды из поваренной соли. Это обеспечило более экономичному аммиачно-содовому производству всеобщее признание и способствовало его промышленному распространению [14, с. 76-78]. Однако в рассматриваемый период ведущую роль в промышленности по-прежнему играл леблановский процесс производства соды. Аммиачный способ развивался, не вытесняя леблановский метод, а параллельно с ним. Этому способствовало расширение емкости рынка содовых продуктов.
В России первый завод начал работать по аммиачно-содовому циклу в 1868 г. Это был Камский содовый завод Лихачева в Казанской губернии. В налаживании его работы принимал участие известный русский химик профессор М. Я. Киттары. Предприятие было рассчитано на выпуск 50 000 пудов кальцинированной соды в год. Продукция завода экспонировалась в 1870 г. на Всероссийской мануфактурной выставке. Предприятие работало 4 года, после чего было закрыто из-за высоких цен в России на поваренную соль.
Изменения в технологии производства серной кислоты. Новые химические продукты
По мере роста масштабов производства соды и других продуктов содовых заводов соответственно росла их потребность в серной кислоте, которая сделалась важнейшим химическим полупродуктом, необходимым в производстве солей п кислот, удобрений и взрывчатых веществ. Уровень производства серной кислоты как бы стал мерилом технико-промышленного потенциала той или иной страны [19, 20].
Для второй половины XVIII в. и почти всего XIX в. характерно распространение камерного способа получения серной кислоты. Первое описание получения концентрированной серной кислоты, называемой «купоросным маслом», дано итальянцем В. Бирингуччо в книге «О пиротехнике» (1540 г.). Сведения о «купоросном масле» содержатся также в трудах немецкого алхимика XV в., известного под именем Василия Валентина (изданные в конце XVI - начале XVII в.)
Несмотря на то что купоросное масло, получаемое из серы, а также из железного купороса, было известно давно, спрос на него был небольшой. Лишь в XVIII в. с развитием капиталистической промышленности появились признаки оживления производства и потребления купоросного масла. Практическое использование серной кислоты началось с введением индиго в 40-х годах XVIII в. для крашения шерсти в Германии. В 50-х годах производство купоросного масла в Германии увеличивается главным образом за счет числа мелких предприятий с численностью работающих на каждом из них от одного до трех человек. Таких предприятий насчитывалось в 90-х годах XVIII в. свыше тридцати лишь в Саксонских рудных горах, которые вырабатывали из 5000 центнеров купороса до 120 000 фунтов купоросного масла [11, с. 186].
Производство купоросного масла существовало в это время во многих районах и городах Германии и в некоторых других европейских странах. Суть способа получения (по Г. Фестеру) состояла в следующем. Выветрившиеся остатки квасцовых сланцев, ранее служивших для получения серы, или огарки пиритсодержащих каменных и бурых углей, выгцелачи-вали. Щелок выпаривали и прокаливали до превращения в сырой купоросный камень, представляющий смесь сернокислых соединений железа и алюминия. Полученный полупродукт загружали в небольшие глиняные реторты, которые помещали по 30 штук одновременно в сильно разогретую галерную перегонную печь: в результате купоросное масло скоплялось в приемниках. Каждая загрузка реторт давала приблизительно по полтора фунта кислоты. Процесс перегонки одной загрузки продолжался 7-8 дней [11, с. 187-188].
Первому промышленному способу получения серной кислоты, положенному в основу камерного процесса, предшествовали работы французских химиков Н. Лемери и Н. Лефевра, предложивших в 1666 г. (по другим данным - в 1690 г.) окислять серу, нагревая ее в смеси с селитрой [21, с. 33]. Указанный путь получения серной кислоты был впервые реализован в заводском масштабе в Англии в 1740 г. По этому методу смесь серы и селитры сжигали в ковше, подвешенном в стеклянном баллоне, содержащем небольшое количество воды. При сжигании выделялся серный ангидрид, который реагировал с водой с образованием серной кислоты.
В 1746 г. Дж. Робук из Бирмингама положил начало камерному способу производства серной кислоты, заменив стеклянные баллоны свинцовыми камерами. В этом процессу серу предварительно смешивали с селитрой, подавали в камеру в небольших железных тележках и сжигали [11, с. 188]. Первый сернокислотный завод был пущен в 1749 г. Дж. Робуком в Англии. Второй сернокислотный завод был выстроен в Престонпансе (Шотландия). Этот завод насчитывал в 1813 г. не менее 108 небольших свинцовых камер. Один из английских заводов даже был оборудован 360 камерами. Серная кислота, изготовлявшаяся по камерному способу, стала поступать на международный рынок и получила название английской [11, с. 189; 21, с. 49].
В течение второй половины XVIII в. в Англии возникли новые сернокислотные заводы. Около одного лишь Глазго работало 6-8 заводов, 8 предприятий было построено в Бирмингаме и его окрестностях. Появление такого числа новых заводов вызывалось постоянно увеличивающимся спросом на серную кислоту со стороны различных потребителей, среди которых большое место занимало текстильное производство. Еще в 1750 г. доктор Хом в Эдинбурге установил, что серную кислоту можно с успехом применять вместо кислого молока для подкисления отбеливаемых льняных и хлопчатобумажных тканей; в результате продолжительность этой операции сократилась с 2-3 недель до 12 ч. Потребность в серной кислоте еще больше возросла, когда был введен процесс отбелки хлором. В 1815 г. потребление серной кислоты в Англии составляло около 3 тыс. т.
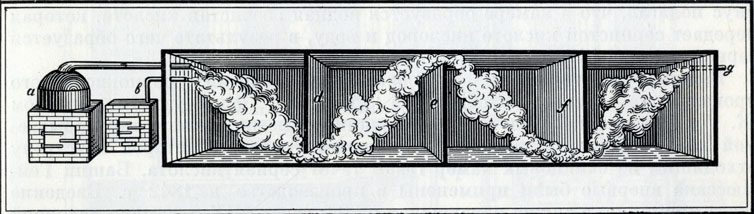
Камерная сернокислотная система начала 30-х годов XIX в. а - паровик; в - серная печь; d, e, f - свинцовые перегородки; g - труба для выпуска газов
По мере развития сернокислотного производства увеличивались размеры камер. Смесь серы и селитры сжигали на железной доске, установленной немного выше уровня жидкости в камере. Сжигание проводили периодически до тех пор, пока удельный вес кислоты не достигал 1,56. После этого кислоту отсасывали с помощью сифонов в свинцовые резервуары и концентрировали сначала в свинцовых, а затем в стеклянных сосудах (при нагревании).
Первое крупное французское предприятие - завод Ж. Голькера в Руане, основанный в 1766 г., сначала работал со стеклянными баллонами, но в 1769 г. здесь были установлены свинцовые камеры. Распространение камерного способа в Германии шло менее успешно; в этой стране преобладал способ получения серной кислоты в стеклянных баллонах. Первая свинцовая камера в Германии появилась в 1812 г. в Швемзале близ Лейпцига, а затем и в некоторых других городах [11, с. 188-190].
В 1805 г. камерный способ производства серной кислоты был введен в России.
С развитием производства серной кислоты начались работы по усовершенствованию технологии камерного процесса. В 1774 г. французский технолог Де-ла Фолье для интенсификации реакции предложил вводить в камеры не воду, а водяной пар, что дало возможность вести процесс непрерывно. Важными оказались выводы, к которым пришли в 1793 г. во Франции Н. Клеман и Ш. Дезорм: они показали, что в камерном процессе сернистый газ окисляется вследствие передачи ему кислорода воздуха окислами азота. В процессе взаимодействия эти окислы восстанавливаются до окиси азота, которая под действием воздуха вновь окисляется, причем этот процесс может идти беспрерывно. В результате оказалось, что расход селитры, выполняющей каталитическую роль переносчика кислорода, можно значительно сократить; кроме того, стала очевидной необходимость замены селитры азотной кислотой. В 1810 г. этот способ был впервые практически реализован во Франции Д. Голькером и вскоре получил всеобщее признание. Серу стали сжигать в отдельной печи, а окислы азота получать разложением селитры серной кислотой [11, с. 188-190; 21, с. 61, 81]. Химические процессы, протекающие в производстве серной кислоты, исследовали Г. Дэви, II. Я. Берцелиус и многие другие ученые. Г. Дэви показал (1812 г.), что химическое взаимодействие между сернистым газом и окисью азота возможно только в присутствии воды и что в безводном состоянии эти газы не взаимодействуют. И. Я. Берцелиус полагал, что в камере образуется водная азотистая кислота, которая передает сернистой кислоте кислород и воду, в результате чего образуется серная кислота [17, с. 34; 22].
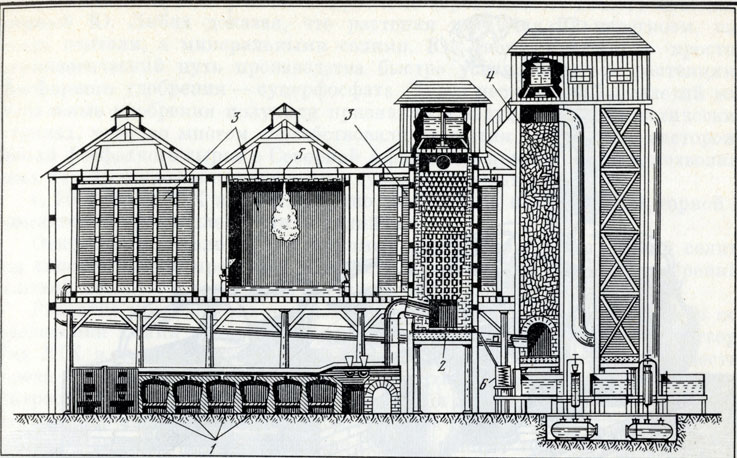
Усовершенствованная заводская камерная сернокислотная установка (конец XIX в.) 1 - печи обжига пирита; 2 - башня Гловера; 3 - свинцовые камеры; 4 - башня Гей-Люссака; 5 - насос; 6 - холодильник
Важным достижением в области технологии сернокислотного производства явилось изобретение (1827 г.) французским химиком Ж. Гей-Люссаком башни для улавливания окислов азота - так называемой холодной башни, наполненной кусками кокса, в которой навстречу отходящим из свинцовых камер газам течет серная кр!слота. Башни Гей-Люссака впервые были применены в производстве в 1842 г. Введение этого принципа, а также начавшаяся в 30-х годах XIX в. замена исходной серы колчеданами значительно снизили стоимость серной кислоты и интенсифицировали процесс [23].
Появление и распространение процесса обжига колчеданов как исходного сырья для получения серной кислоты - важный этап в истории сернокислотного производства. Замена самородной серы колчеданами - сернистыми соединениями железа (пирит), меди, свинца, цинка, сурьмы и некоторыми другими - была вызвана резким повышением цен на сицилийскую серу, потребление и цены на которую с развитием сернокислотного и порохового производства неустанно росли. В 1838 г. цены на сицилийскую серу возросли в 2,8 раза [16, с. 43; 24].
Под влиянием повышения цен на серу поиски новой технологии развернулись почти одновременно в нескольких странах Европы. В 1833 г. французская фирма «Перре и сын» (Лион) ввела на своем производстве обжиг пиритов, отказавшись тем самым от серы [25, с. 14]. К 1837 г. относится начало использования колчеданов в производстве серной кислоты в Австрии, а в 1838 г. - в Англии, где новая технология стала быстро развиваться. Менее чем через год в Англии было получено 15 привилегий на способы получения серной кислоты из колчеданного сырья. Но имеются сведения, что до этого в 1817 г. н Дептфорде технолог Гилль употреблял серный колчедан вместо серы в качестве сырья для получения серной кислоты. В результате внедрения новой технологии в Германии (Саксония) впервые в этой стране в 1840 г. появилась так называемая металлургическая серная кислота, получаемая обжигом медного колчедана, свинцового блеска и цинковой обманки [15; 17, с. 447]. Это наглядный пример начала комбинированного развития металлургического и химического производств, которое получило широчайшее распространение в более поздний период.
Дальнейшим шагом в усовершенствовании сернокислотного производства явилось создание (1859 г.) английским технологом Дж. Гловером так называемой горячей башни, в которой нагретый сернистый газ, образующийся при обжиге сернистых руд, поступает навстречу кислоте, содержащей окислы азота в связанном виде (нитрозилсерная кислота). Часть сернистого газа окисляется в башне Гловера до серного ангидрида, который поглощается серной кислотой. Выделяющиеся из нитро-зилсерной кислоты (известной ранее как «камерные кристаллы») окислы азота и газы, еще содержащие сернистый газ, поступают последовательно в ряд камер, в которые одновременно впускают пар. В камерах сернистый газ окисляется до серного ангидрида и, соединяясь с водой, образует серную кислоту. Отработанные окислы азота поступают из камер в одну или несколько башен Гей-Люссака, где поглощаются стекающей вниз серной кислотой с образованием нитрозилсерной кислоты, вновь идущей в производство.
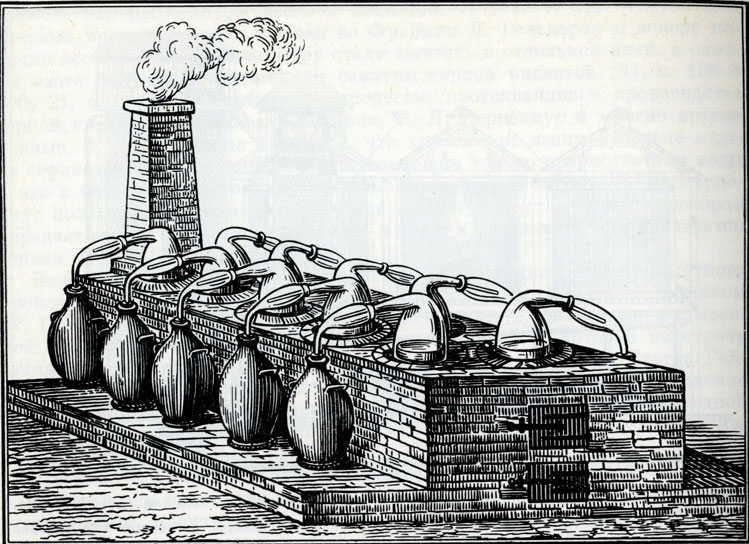
Сгущение серной кислоты в стеклянных ретортах (XIX в.)
Наряду с камерным способом производства серной кислоты в конце XIX в. начал развиваться контактный процесс. Принцип контактного способа получения серной кислоты был открыт в 1831 г. П. Филипсом (Англия), внесшим предложение окислять сернистый ангидрид непосредственно кислородом воздуха при пропускании газовой смеси через накаленный платиновый катализатор [17, с. 42; 26].
Первый завод контактного производства серной кислоты был пущен в 1847 г. (Бельгия), но вскоре был закрыт. Вплоть до 70-х годов XIX - начала XX в. контактный метод не получил практического развития в связи с тем, что не удавалось точно установить причины отравления платинового катализатора и не были установлены физико-химические факторы, влияющие на процесс.
Кроме того, спрос на олеум (серная кислота, содержащая избыток серного ангидрида - до 20%) был невелик. Для получения олеума наиболее целесообразным был контактный процесс. Потребность в олеуме возросла лишь в 70-x годах XIX в., когда развилась промышленность синтетических красителей и других химических производств.
Контактный метод производства серной кислоты начал развиваться благодаря работам К. Винклера в середине 70-х годов XIX в., а затем шь лучил широкое распространение в начале XX в. после выявления Р. Книтчем (Германия) причин отравления катализатора в промышленных условиях, разработки методов очистки сернистого газа от вредных примесей, а также установления основных физико-химических закономерностей протекания процесса.
Крепкую серную кислоту на заводах часто получали сгущением в стеклянных ретортах, нагреваемых на специальных печах. Шейку каждой реторты вставляли в отверстие изогнутой стеклянной насадки, нижний конец которой опущен в глиняный или стеклянный сосуд, служащий приемником для выделяющейся воды.
В 1867 г. производство серной кислоты в экономически развитых капиталистических странах составило, т: Англия-155 тыс., Франция - 125 тыс., Германия - 75 тыс., США - 5 тыс., Австро-Венгрия - 15 тыс Бельгия - 20 тыс. Мировое производство в 1878 г. достигло почти 1 млн. т
Прогресс в производстве серной кислоты способствовал созданию удобрений для сельского хозяйства. 30-40-е годы XIX в. ознаменовались повышенным интересом ученых к проблеме агрохимии. В 1840 г. немецкий ученый Ю. Либих доказал, что растения питаются не перегноем, каь тогда считали, а минеральными солями. Ю. Либих предложил простой технологический путь производства быстро усваивающегося растениями фосфорного удобрения - суперфосфата. Через несколько десятилетий минеральные удобрения получили признание в ведущих капиталистических странах, чему во многом способствовали открытия богатейших месторождений фосфатного сырья в Северной Африке и в других странах, позволивших начать широкое строительство суперфосфатных заводов.
С 70-х годов XIX в. начала бурно развиваться новая отрасль горной и химической промышленности - калийная.
Открытие в начале XIX в. крупных месторождений чилийской селитры способствовало прогрессу в области производства азотных удобрений, азотной кислоты и взрывчатых веществ.
Развитие производства азотной кислоты и широко развернувшиеся исследования химии нитросоединений привели во второй и третьей четвертях XIX в. к открытию ряда сильно действующих взрывчатых веществ. Среди них особое значение имели бризантные взрывчатые вещества - нитроглицерин и пироксилин, оказавшие огромное влияние на военную технику и горнодобывающую промышленность.
Заложенные в первой половине XIX в. основы тонкого органического синтеза получили еще большее развитие с 50-х годов прошлого столетия, в результате чего был синтезирован ряд ароматических углеводородов, красителей (ализарин, индиго, фуксин и др.), лекарственных и косметических препаратов. На основе этих работ возникла крупная анилинокра-сочная и фармацевтическая промышленвость [27].
Из других работ по синтезу, выполненных в середине XIX в. и оказавших влияние на прогресс технологии, необходимо отметить следующие: синтез уксусной кислоты, электросинтез углеводородов, синтезы метилового спирта, муравьиной кислоты, бензола и ряд других [18].
Крупные достижения в области химии и химической технологии характеризуют качественно новый уровень, которого достигло химическое производство после промышленной революции конца XVIII - начала XIX в.
|
ПОИСК:
|
При использовании материалов сайта активная ссылка обязательна:
http://nplit.ru/ 'Библиотека юного исследователя'